Overview
Gaucher (go-SHAY) disease is the result of a buildup of certain fatty substances in certain organs, particularly your spleen and liver. This causes these organs to enlarge and can affect their function.
The fatty substances also can build up in bone tissue, weakening the bone and increasing the risk of fractures. If the bone marrow is affected, it can interfere with your blood's ability to clot.
An enzyme that breaks down these fatty substances doesn't work properly in people with Gaucher disease. Treatment often includes enzyme replacement therapy.
An inherited disorder, Gaucher disease is most common in Jewish people of Eastern and Central European descent (Ashkenazi). Symptoms can appear at any age.
Products & Services
Symptoms
There are different types of Gaucher disease, and signs and symptoms of disease vary widely, even within the same type. Type 1 is by far the most common.
Siblings, even identical twins, with the disease can have different levels of severity. Some people who have Gaucher disease have only mild or no symptoms.
Most people who have Gaucher disease have varying degrees of the following problems:
- Abdominal complaints. Because the liver and especially the spleen can enlarge dramatically, the abdomen can become painfully distended.
- Skeletal abnormalities. Gaucher disease can weaken bone, increasing the risk of painful fractures. It can also interfere with the blood supply to your bones, which can cause portions of the bone to die.
- Blood disorders. A decrease in healthy red blood cells (anemia) can result in severe fatigue. Gaucher disease also affects the cells responsible for clotting, which can cause easy bruising and nosebleeds.
More rarely, Gaucher disease affects the brain, which can cause abnormal eye movements, muscle rigidity, swallowing difficulties and seizures. One rare subtype of Gaucher disease begins in infancy and typically results in death by 2 years of age.
When to see a doctor
If you or your child has the signs and symptoms associated with Gaucher disease, make an appointment with your doctor.
Causes
Autosomal recessive inheritance pattern
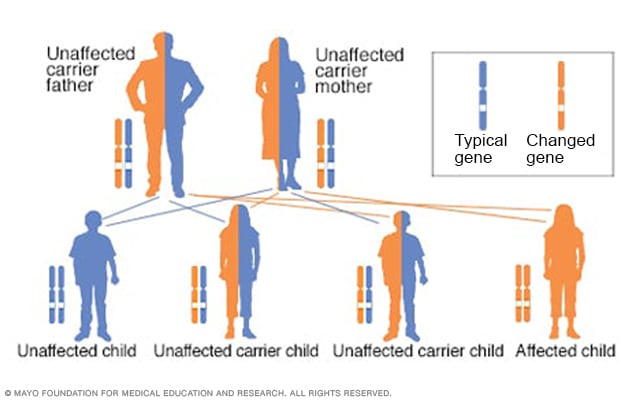
Autosomal recessive inheritance pattern
To have an autosomal recessive disorder, you inherit two changed genes, sometimes called mutations. You get one from each parent. Their health is rarely affected because they have only one changed gene. Two carriers have a 25% chance of having an unaffected child with two unaffected genes. They have a 50% chance of having an unaffected child who also is a carrier. They have a 25% chance of having an affected child with two changed genes.
Gaucher disease is passed along in an inheritance pattern called autosomal recessive. Both parents must be carriers of a Gaucher changed (mutated) gene for their child to inherit the condition.
Risk factors
People of Eastern and Central European Jewish (Ashkenazi) ancestry are at higher risk of developing the most common variety of Gaucher disease.
Complications
Gaucher disease can result in:
- Delays in growth and puberty in children
- Gynecological and obstetric problems
- Parkinson's disease
- Cancers such as myeloma, leukemia and lymphoma