Overview
Medium-chain acyl-coenzyme A dehydrogenase (MCAD) deficiency is an inherited disorder that prevents your body from breaking down certain fats and turning them into energy. Your metabolism involves the processes your body uses to produce energy. MCAD deficiency can cause problems with your metabolism.
If left untreated, MCAD deficiency may lead to severe lack of energy and tiredness, liver disease, coma, and other serious health problems. Also, the level of sugar in your blood can drop dangerously low — a condition called hypoglycemia. Prevention and prompt treatment are essential regardless of blood sugar level. If you have MCAD deficiency, a sudden episode, called a metabolic crisis, can be caused by common illnesses, high fever, stomach problems or going too long without eating, known as prolonged fasting.
MCAD deficiency is present from birth and is a lifelong condition. In the United States, all states test for MCAD deficiency at birth as part of newborn screening. Many other countries also provide routine newborn screening for MCAD deficiency. If MCAD deficiency is diagnosed and treated early, the disorder can be well managed through diet and lifestyle.
Symptoms
MCAD deficiency is usually first identified in babies and young children. In rare cases, the disorder is not diagnosed until adulthood.
Symptoms can vary among people with MCAD deficiency. They may include:
- Vomiting.
- Low or no energy.
- Weak feeling.
- Low blood sugar.
A sudden severe episode, called a metabolic crisis, can be due to:
- Going too long without eating.
- Missing meals.
- Common infections.
- High fever.
- Ongoing stomach and digestive problems that may cause vomiting and diarrhea.
- Intense exercise.
When to see a doctor
In the United States and many other countries, newborn screening programs test for MCAD deficiency. After your first evaluation, you may be referred to a specialist in evaluating and treating MCAD deficiency. You may also be referred to other health care team members, such as a registered dietitian.
Contact your health care team if you've been diagnosed with MCAD deficiency and have a high fever, no appetite, stomach or digestive symptoms, or a planned medical procedure that requires fasting.
Causes
Autosomal recessive inheritance pattern
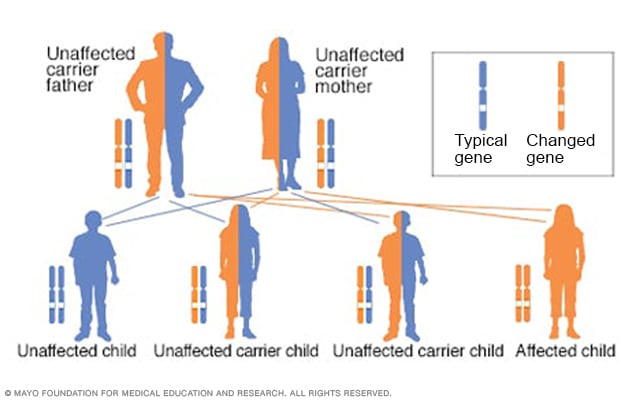
Autosomal recessive inheritance pattern
To have an autosomal recessive disorder, you inherit two changed genes, sometimes called mutations. You get one from each parent. Their health is rarely affected because they have only one changed gene. Two carriers have a 25% chance of having an unaffected child with two unaffected genes. They have a 50% chance of having an unaffected child who also is a carrier. They have a 25% chance of having an affected child with two changed genes.
When you don't have enough of the MCAD enzyme in your body, certain fats called medium-chain fatty acids can't be broken down and turned into energy. This leads to low energy and low blood sugar. Also, fatty acids can build up in body tissues and cause damage.
MCAD deficiency is caused by a change in the ACADM gene. The condition is inherited from both parents in an autosomal recessive pattern. This means that both parents are carriers — each has one changed gene and one unchanged gene — but they don't have symptoms of the condition. The child with MCAD deficiency inherits two copies of the changed gene — one from each parent.
If you inherit only one changed gene, you won't develop MCAD deficiency. With one changed gene, you are a carrier and can pass the changed gene to your children. But they wouldn't develop the condition unless they also inherited a changed gene from their other parent.
Risk factors
A child is at risk of MCAD deficiency if both parents are carriers of a gene known to cause it. The child then inherits two copies of the gene — one from each parent. Children who inherit only one copy of the affected gene from one parent typically won't develop MCAD deficiency but will be carriers of the gene.
Complications
If metabolic crisis caused by MCAD deficiency is left untreated, it can lead to:
- Seizures.
- Liver problems.
- Brain damage.
- Coma.
- Sudden death.
Nov. 28, 2023